Loading 'brms' package (version 2.21.0). Useful instructions
can be found by typing help('brms'). A more detailed introduction
to the package is available through vignette('brms_overview').
Attaching package: 'brms'
The following object is masked from 'package:stats':
ar
To be able to fit an animal model, brms needs the relativeness (relationship) matrix of the pedigree and not its inverse (as in other softwares). This can be estimated using the nadiv package created by Pr. Matthew Wolak (https://cran.r-project.org/web/packages/nadiv/index.html).
We are now ready to specify our first model: The structure of a bmrs model is similar to lme4, thus the random effect is added to the model with the term (1 | gr(animal, cov = Amat) which associate the id animal to the matrix of relativeness. In addition to the synthase of lme4, we includes other features or parameters within the models such as chain which represent the number of Markov chains (defaults to 4), core which represents the number of cores to use when executing the chains in parallel and iter which represents the number of total iterations per chain. For more parameters such as thin or warmup/burnin, you can read the Cran R page of the package (https://cran.r-project.org/web/packages/brms/brms.pdf)
bmrs is a Bayesian Multilevel Models using Stan, doing so we can apply a prior to the model to better shape the distribution of the different variances estimated by the model. Given that bmrs fit the model using a Bayesian approach via the software stan, we need to specify priors for the model. Default priors in brms work relatively well, however we strongly suggest to carefully select an adequate prior for your analysis. In this tutorial we will use the default priors. To get the prior used by default, we can use the get_prior() function.
brms_m1.1<-brm(bwt~1+(1|gr(animal, cov =Amat)), data =gryphon, data2 =list(Amat =Amat), family =gaussian(), chains =1, cores =1, iter =100)save(brms_m1.1, file ="r-obj/brms_m1_1.rda")
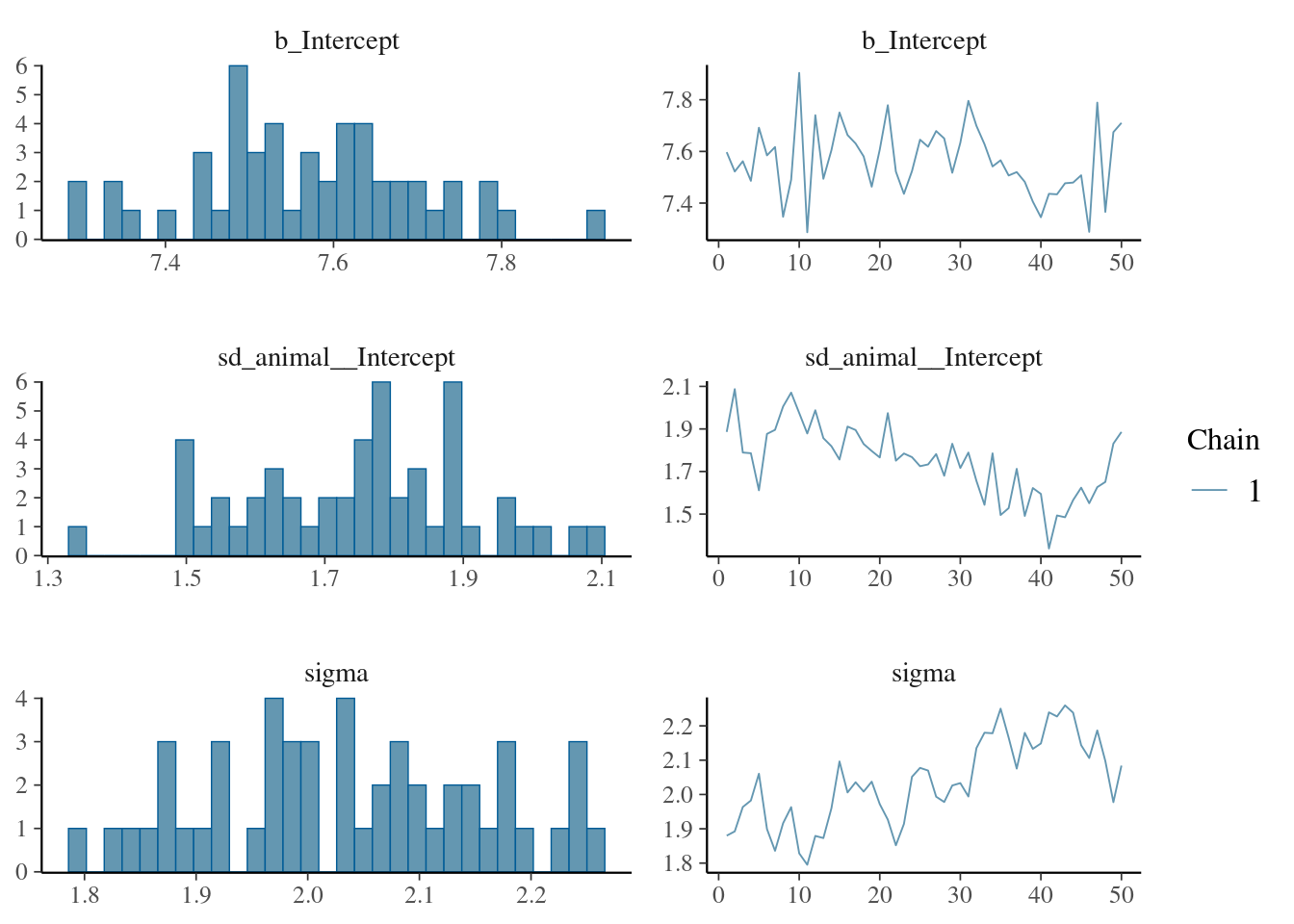
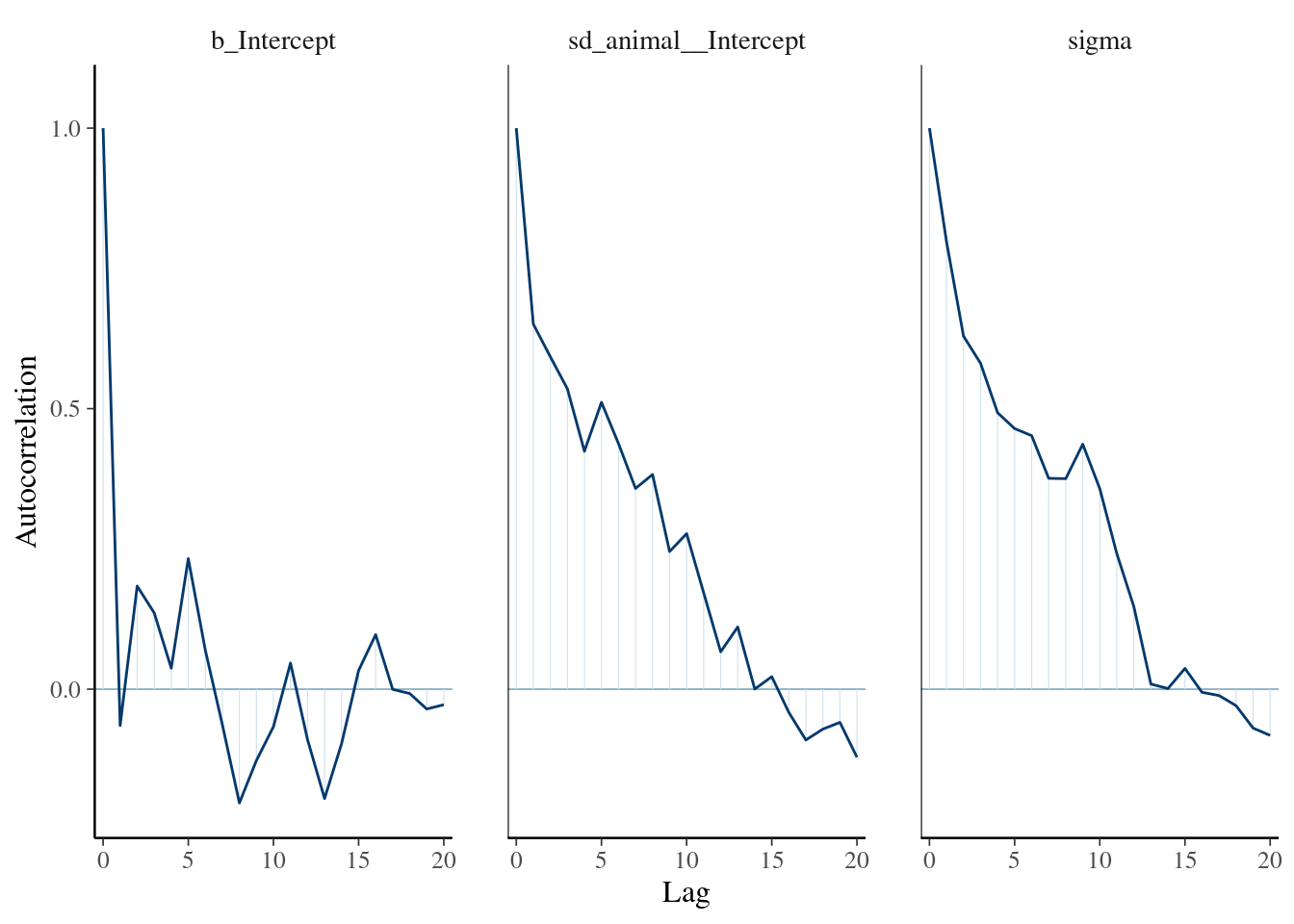
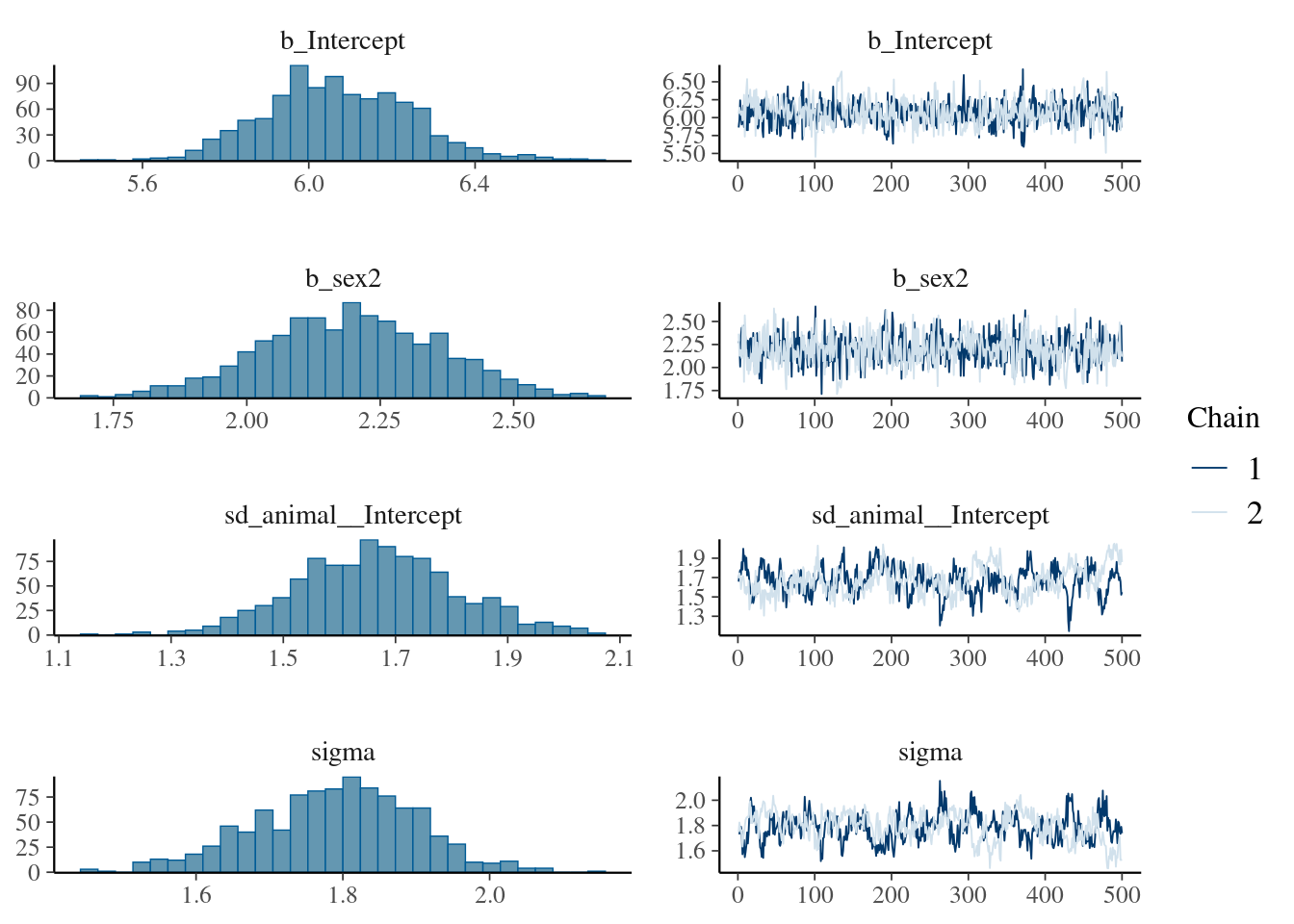
The result of the long model calculation is save in a spare file brms_m1_1.rda". To help readers, we can directly reloading it. Two distinct plot can be produce to produce some diagnostics graphs mcmc_plot.Note, that sigma represents the residual standard deviation.
Next,we examine (or directly using the model) the variance estimate and their distributions (via summary or plot).
Warning: Parts of the model have not converged (some Rhats are > 1.05). Be
careful when analysing the results! We recommend running more iterations and/or
setting stronger priors.
Family: gaussian
Links: mu = identity; sigma = identity
Formula: bwt ~ 1 + (1 | gr(animal, cov = Amat))
Data: gryphon (Number of observations: 854)
Draws: 1 chains, each with iter = 100; warmup = 50; thin = 1;
total post-warmup draws = 50
Multilevel Hyperparameters:
~animal (Number of levels: 1084)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sd(Intercept) 1.75 0.17 1.49 2.06 1.55 2 20
Regression Coefficients:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept 7.57 0.13 7.30 7.79 1.00 27 63
Further Distributional Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 2.04 0.12 1.83 2.25 1.68 2 20
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
The plot of variance showed that the different variances have an normal distribution, the autocorrelation plot or ‘acf’ show that the autocorrelation is close to 0. The summary exposes the mean (Estimate) of each variance or fixed effect (here just the intercept) associated to their posterior distribution with standard deviation (Est.Error) and two-sided 95% Credible intervals. Rhat provides information on the estimate convergence. If it’s greater than 1, the chains have not yet converged and it will be require to run more iterations and/or set stronger priors. ESS represents the Effective sample values as the number of independent samples from the posterior distribution. However, for the purpose of this guide, the Rhat values are acceptable.
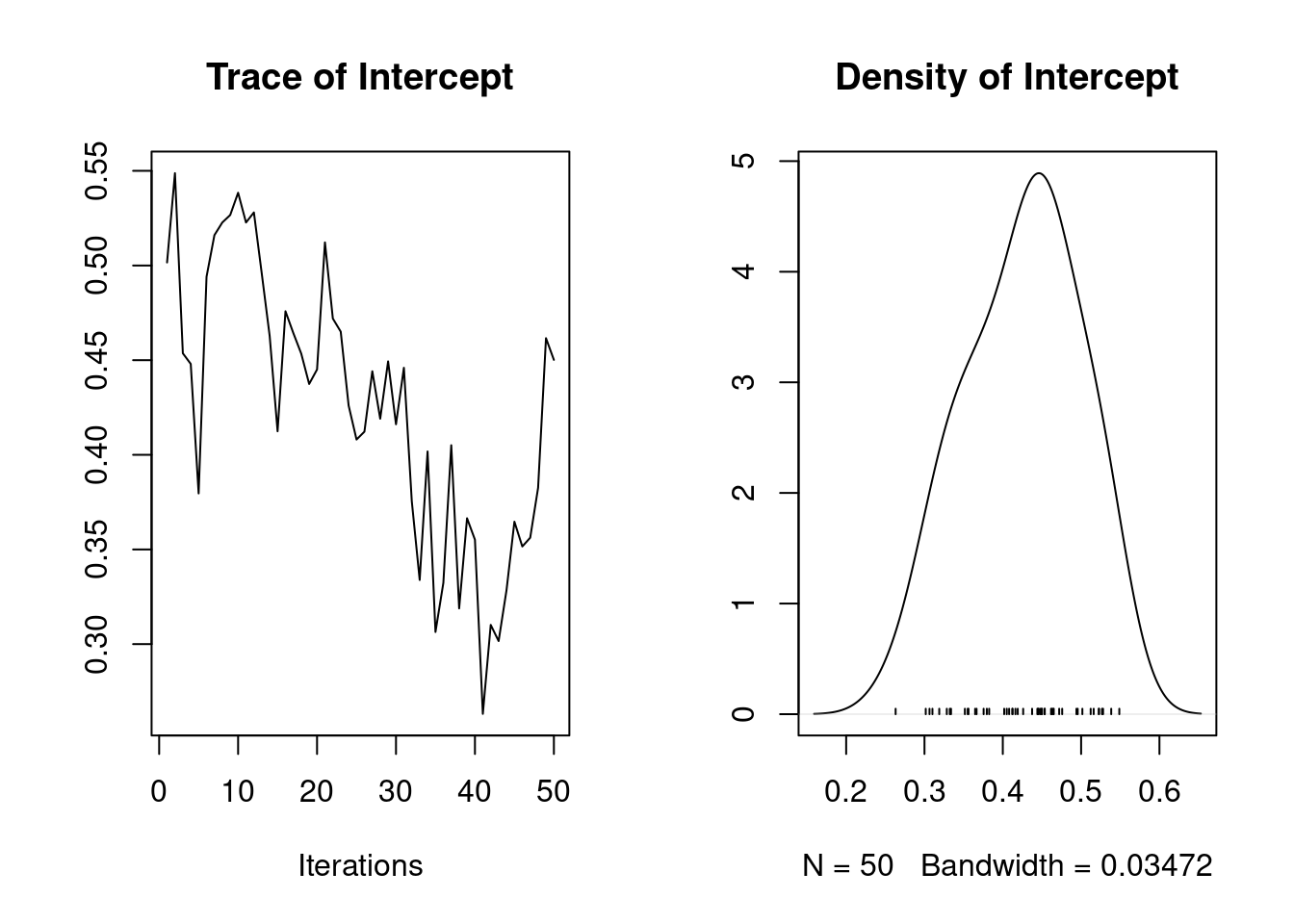
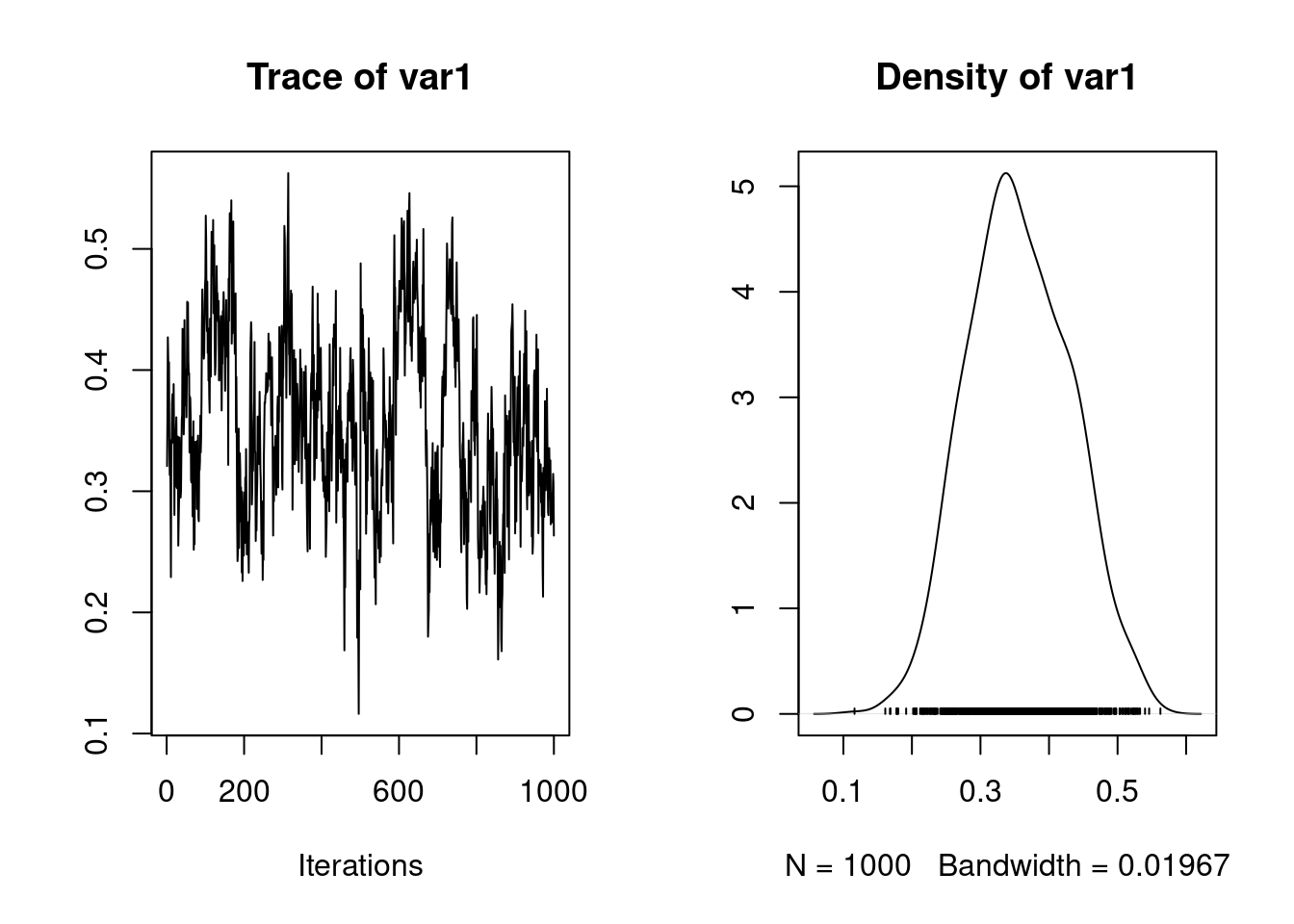
It is also possible to calculate the heritability using the function ‘as.mcmc’
Iterations = 1:50
Thinning interval = 1
Number of chains = 1
Sample size per chain = 50
1. Empirical mean and standard deviation for each variable,
plus standard error of the mean:
Mean SD Naive SE Time-series SE
0.42526 0.07162 0.01013 0.02854
2. Quantiles for each variable:
2.5% 25% 50% 75% 97.5%
0.3027 0.3687 0.4408 0.4702 0.5361
Iterations = 1:50
Thinning interval = 1
Number of chains = 1
Sample size per chain = 50
1. Empirical mean and standard deviation for each variable,
plus standard error of the mean:
Mean SD Naive SE Time-series SE
0.42526 0.07162 0.01013 0.02854
2. Quantiles for each variable:
2.5% 25% 50% 75% 97.5%
0.3027 0.3687 0.4408 0.4702 0.5361
To add effects to a univariate model, we simply modify the priors and the fixed effect portion of the model specification:
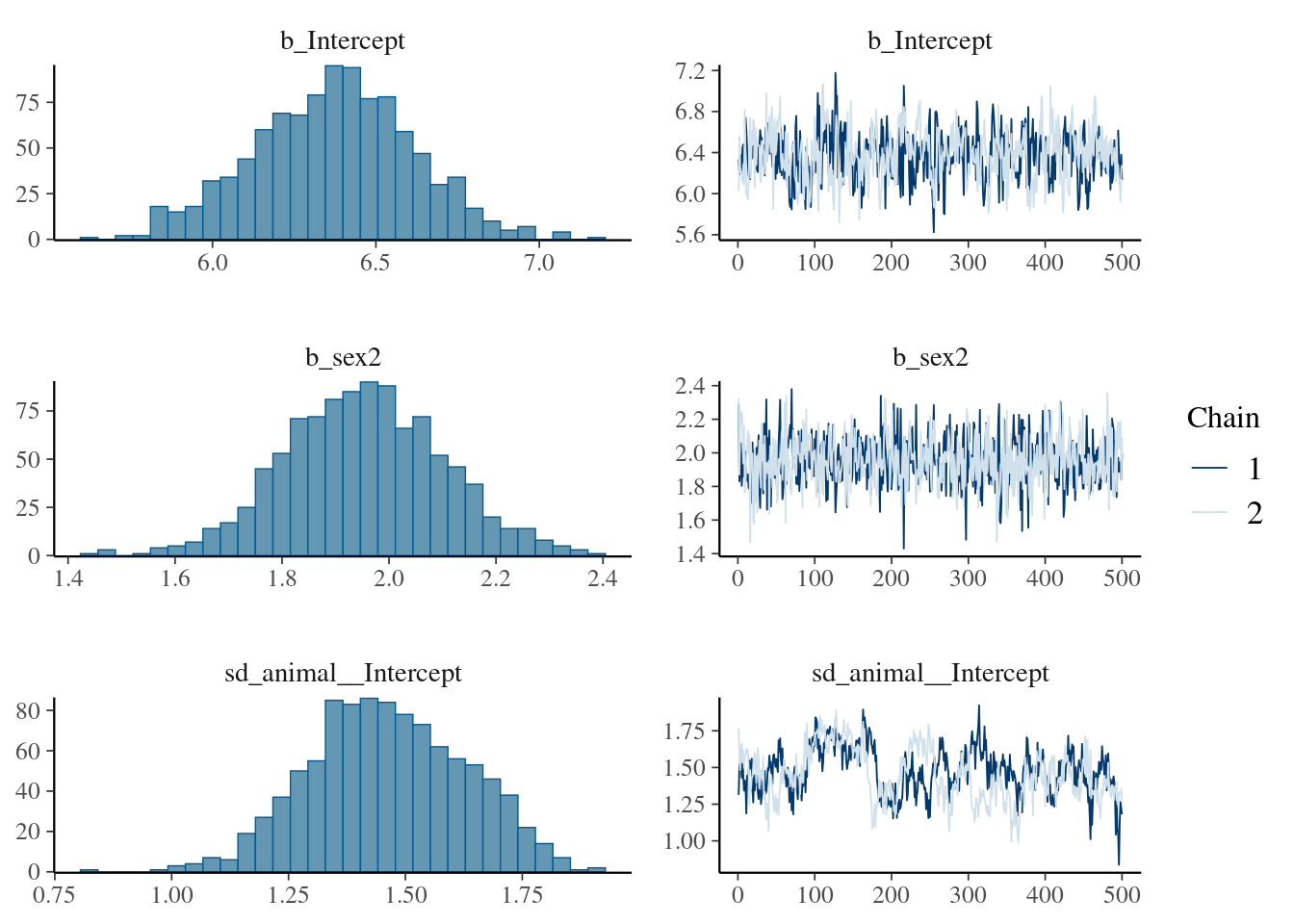
brms_m1.2<-brm(bwt~1+sex+(1|gr(animal, cov =Amat)), data =gryphon, data2 =list(Amat =Amat), family =gaussian(), chains =2, cores =2, iter =1000)save(brms_m1.2, file ="r-obj/brms_m1_2.rda")
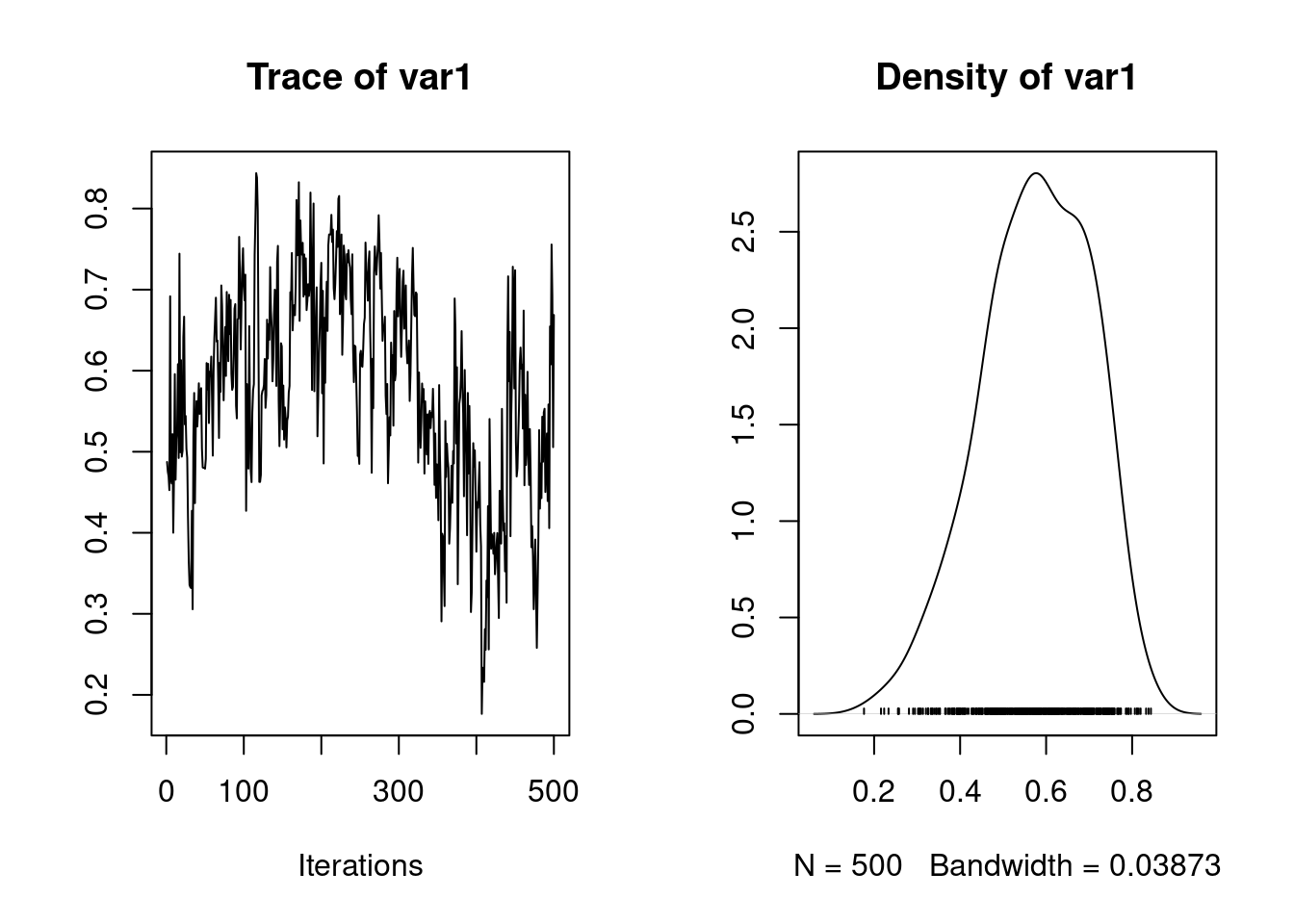
To save time, the results of the calculation is stored in the spare file brms_m1_2.rda". We can assess the significance of sex as a fixed effect by examining its posterior distribution.
$animal
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sd(Intercept) 1.667668 0.1442392 1.393627 1.967308 1.023006 93.37621 119.8231
The posterior distribution of the sex2 term does not overlap zero. Thus, we can infer that sex has an effect on birth weight (presence of a sexual dimorphism) in this model and is a useful addition to the model, for most purposes. It is also worth noting that the variance components have changed slightly:
$animal
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sd(Intercept) 1.667668 0.1442392 1.393627 1.967308 1.023006 93.37621 119.8231
In fact since sex effects were previously contributing to the residual variance of the model our estimate of \(V_R\) (denoted ’units’ in the output) is now slightly lower than before. This has an important consequence for estimating heritability since if we calculate \(V_P\) as \(V_A +V_R\) then as we include fixed effects we will soak up more residual variance driving \(V_P\) . Assuming that \(V_A\) is more or less unaffected by the fixed effects fitted then as \(V_P\) goes down we expect our estimate of \(h^2\) will go up.
Iterations = 1:1000
Thinning interval = 1
Number of chains = 1
Sample size per chain = 1000
1. Empirical mean and standard deviation for each variable,
plus standard error of the mean:
Mean SD Naive SE Time-series SE
0.464637 0.068645 0.002171 0.007561
2. Quantiles for each variable:
2.5% 25% 50% 75% 97.5%
0.3375 0.4183 0.4620 0.5089 0.6030
Iterations = 1:50
Thinning interval = 1
Number of chains = 1
Sample size per chain = 50
1. Empirical mean and standard deviation for each variable,
plus standard error of the mean:
Mean SD Naive SE Time-series SE
0.42526 0.07162 0.01013 0.02854
2. Quantiles for each variable:
2.5% 25% 50% 75% 97.5%
0.3027 0.3687 0.4408 0.4702 0.5361
Here \(h^2\) has increased slightly from 0.5010 to 0.4192 (again, your values may differ slightly due to Monte Carlo error). Which is the better estimate? It depends on what your question is. The first is an estimate of the proportion of variance in birth weight explained by additive effects, the latter is an estimate of the proportion of variance in birth weight after conditioning on sex that is explained by additive effects. An important piece of advice, each researcher should be consistent in how they name their estimates and always correctly describe which estimates they are using conditional or not (to avoid any confusion).
4.0.3 Adding random effects
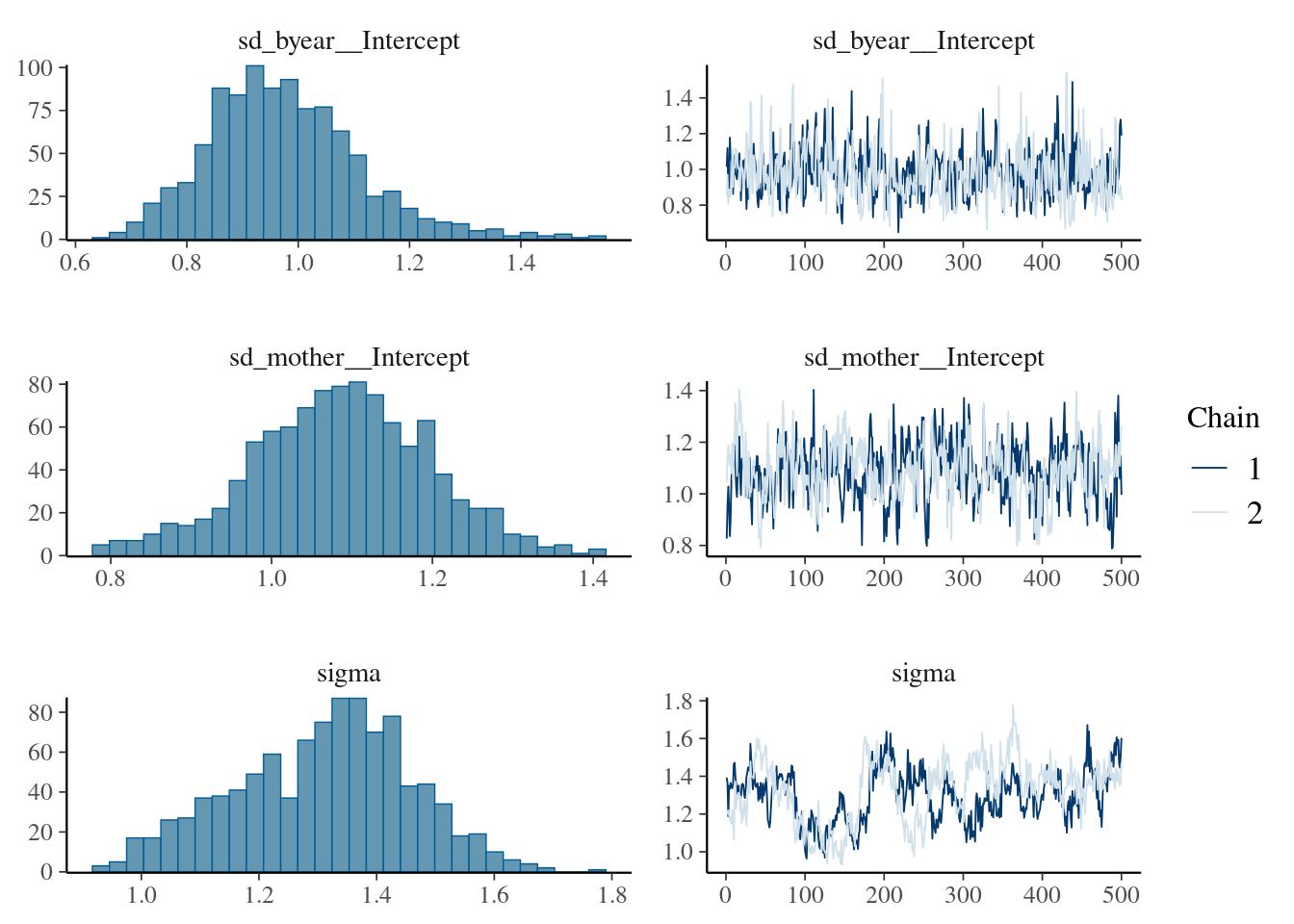
This is done by simply modifying the model statement in the same way, but requires addition of a prior for the new random effect. For instance, we can fit an effect of birth year:
brms_m1.3<-brm(bwt~1+sex+(1|gr(animal, cov =Amat))+(1|byear)+(1|mother), data =gryphon, data2 =list(Amat =Amat), family =gaussian(), chains =2, cores =2, iter =1000)save(brms_m1.3, file ="r-obj/brms_m1_3.rda")
To save time, the results of the calculation is stored in the spare file brms_m1_3.rda". We can assess the significance of sex as a fixed effect by examining its posterior distribution.
load("r-obj/brms_m1_3.rda")plot(brms_m1.3, ask =FALSE, N =3)
Warning: Argument 'N' is deprecated. Please use argument 'nvariables' instead.
Warning: Parts of the model have not converged (some Rhats are > 1.05). Be
careful when analysing the results! We recommend running more iterations and/or
setting stronger priors.
$animal
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sd(Intercept) 1.462757 0.1673822 1.153225 1.768881 1.079817 32.11486 156.8095
$byear
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sd(Intercept) 0.9767418 0.140156 0.7415211 1.306467 1.007016 469.316 407.5334
$mother
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sd(Intercept) 1.086973 0.1107873 0.8507715 1.296683 1.001921 228.2474 380.5216
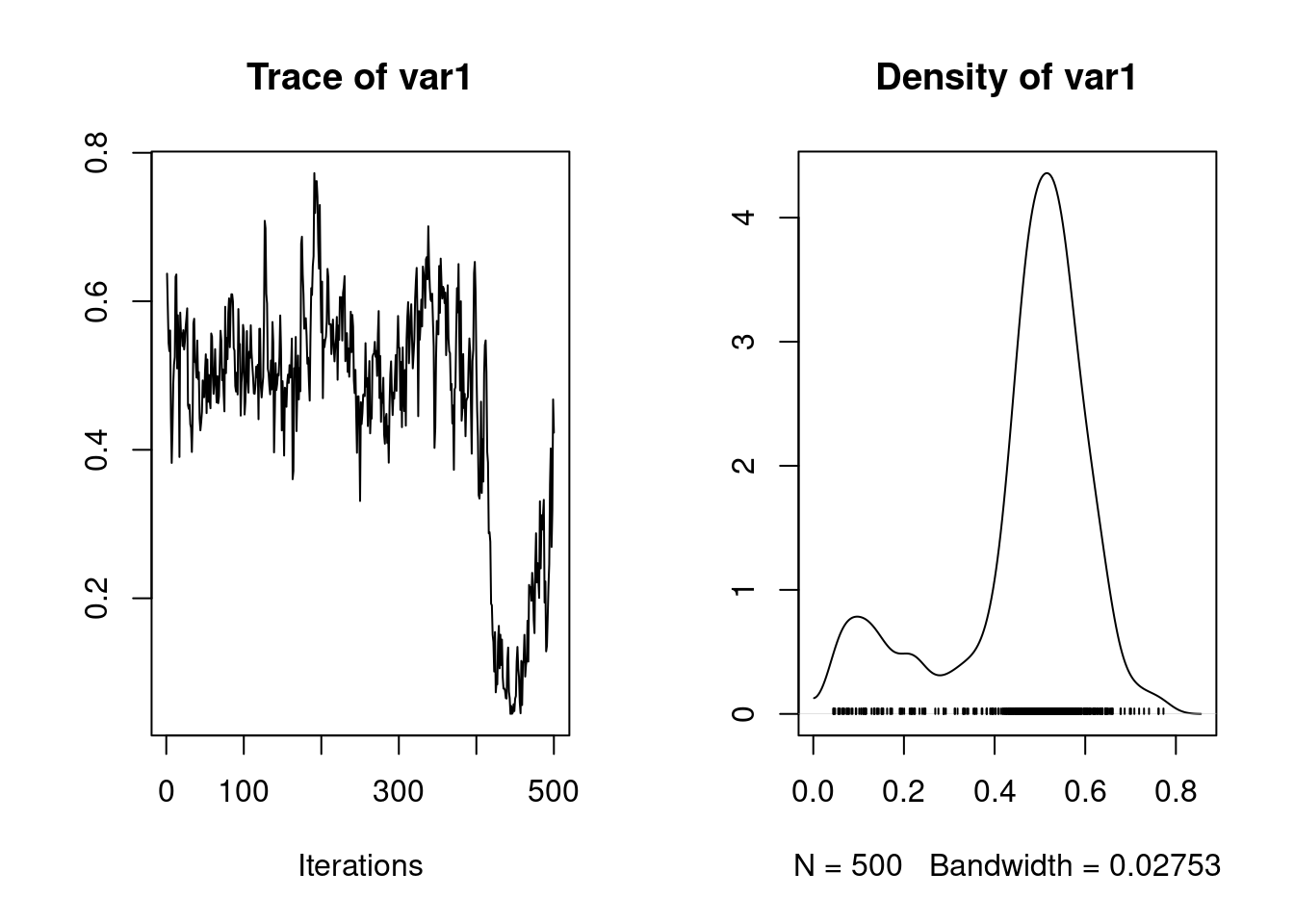
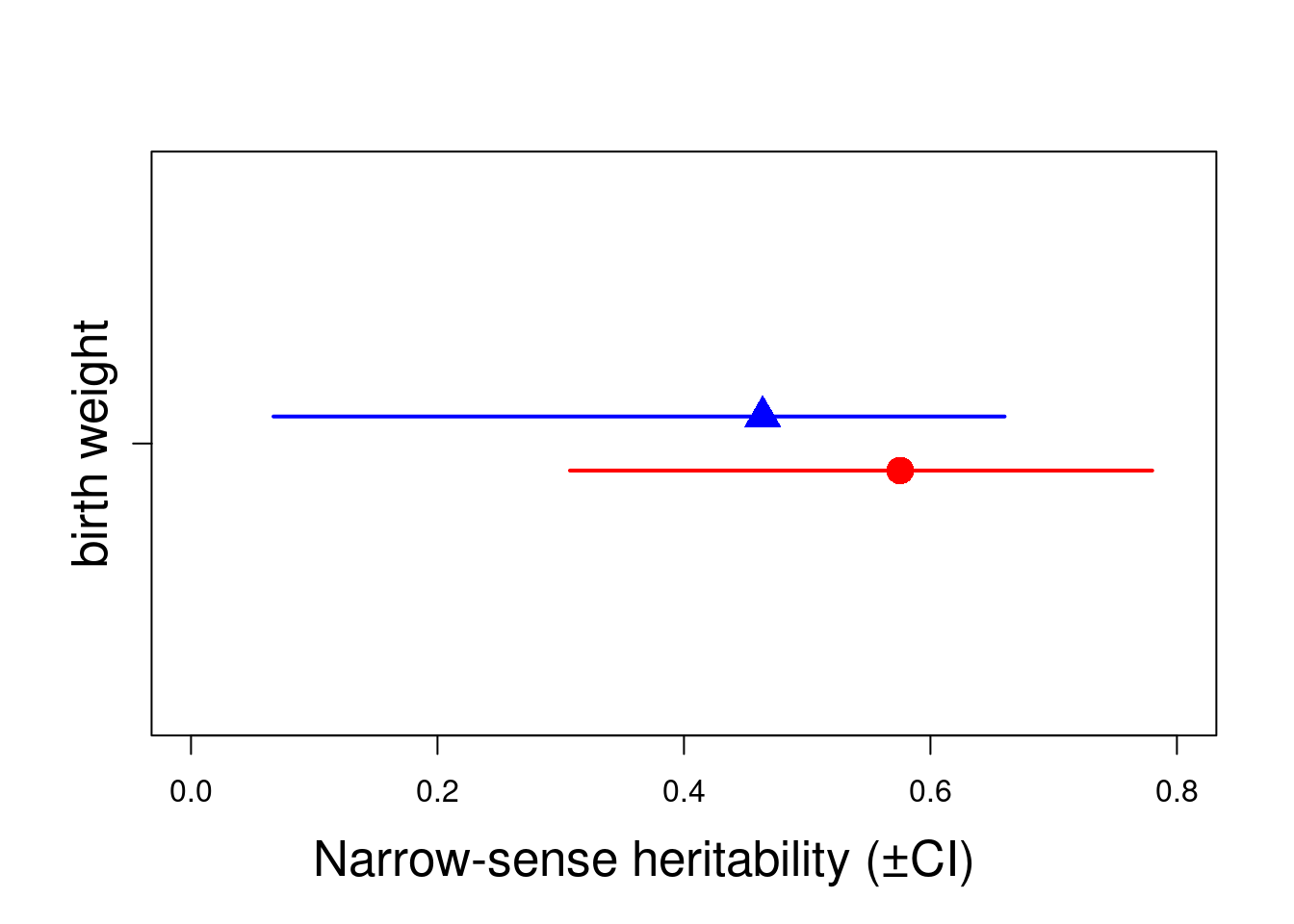
Here partitioning of significant birth year and maternal variance has resulted in a further decrease in \(V_R\) but also a decrease in \(V_A\). The latter is because maternal effects of the sort we simulated (fixed differences between mothers) will have the consequence of increasing similarity among maternal siblings. Consequently they can look very much like an additive genetic effects and if present, but unmodelled, represent a type of ‘common environment effect’ that can - and will- cause upward bias in \(V_A\) and so \(h^2\). Let’s compare the estimates of heritability from each of models 1.2, 1.3 and 1.4:
Iterations = 1:1000
Thinning interval = 1
Number of chains = 1
Sample size per chain = 1000
1. Empirical mean and standard deviation for each variable,
plus standard error of the mean:
Mean SD Naive SE Time-series SE
0.356562 0.073890 0.002337 0.010196
2. Quantiles for each variable:
2.5% 25% 50% 75% 97.5%
0.2208 0.3042 0.3517 0.4111 0.5045
Iterations = 1:1000
Thinning interval = 1
Number of chains = 1
Sample size per chain = 1000
1. Empirical mean and standard deviation for each variable,
plus standard error of the mean:
Mean SD Naive SE Time-series SE
0.464637 0.068645 0.002171 0.007561
2. Quantiles for each variable:
2.5% 25% 50% 75% 97.5%
0.3375 0.4183 0.4620 0.5089 0.6030
Iterations = 1:50
Thinning interval = 1
Number of chains = 1
Sample size per chain = 50
1. Empirical mean and standard deviation for each variable,
plus standard error of the mean:
Mean SD Naive SE Time-series SE
0.42526 0.07162 0.01013 0.02854
2. Quantiles for each variable:
2.5% 25% 50% 75% 97.5%
0.3027 0.3687 0.4408 0.4702 0.5361
Iterations = 1:1000
Thinning interval = 1
Number of chains = 1
Sample size per chain = 1000
1. Empirical mean and standard deviation for each variable,
plus standard error of the mean:
Mean SD Naive SE Time-series SE
0.356562 0.073890 0.002337 0.010196
2. Quantiles for each variable:
2.5% 25% 50% 75% 97.5%
0.2208 0.3042 0.3517 0.4111 0.5045
While testing the significance of fixed effects by evaluating whether or not their posterior distributions overlap zero was simple and valid, this approach does not work for variance components. Variance components are bounded to be positive (given a proper prior), and thus even when a random effect is not meaningful, its posterior distribution will never overlap zero.
Model comparisons can be performed using the function loo_compare using waic or weighted AIC.
Warning: Found 364 observations with a pareto_k > 0.67 in model 'brms_m1.3'. We
recommend to run more iterations to get at least about 2200 posterior draws to
improve LOO-CV approximation accuracy.
Warning: Found 476 observations with a pareto_k > 0.41 in model 'brms_m1.1'. We
recommend to run more iterations to get at least about 2200 posterior draws to
improve LOO-CV approximation accuracy.
Depending of the research question and the presence of different group within the dataset, brms allowed to partition the variance at different groups. Two distinct approch can be done to partition the different random effect: using an extra argument by=sex or by adding (0+sex|) before the |. Notes, here we used || which not estimate a possible covariance between groups (female and male) for the random effect.
brms_m1.4<-brm(# bwt ~ 1 + sex + (1 | gr(animal, cov = Amat, by = sex))+ (1 | gr(byear, by = sex)) + (1 | gr(mother, by = sex)),bwt~1+sex+(0+sex||gr(animal, cov =Amat))+(0+sex||byear)+(0+sex||mother), data =gryphon, data2 =list(Amat =Amat), family =gaussian(), chains =2, cores =2, iter =1000)save(brms_m1.4, file ="r-obj/brms_m1_4.rda")
To save time, the results of the calculation is stored in the spare file brms_m1_4.rda".
Warning: Parts of the model have not converged (some Rhats are > 1.05). Be
careful when analysing the results! We recommend running more iterations and/or
setting stronger priors.
Family: gaussian
Links: mu = identity; sigma = identity
Formula: bwt ~ 1 + sex + (0 + sex || gr(animal, cov = Amat)) + (0 + sex || byear) + (0 + sex || mother)
Data: gryphon (Number of observations: 854)
Draws: 2 chains, each with iter = 1000; warmup = 500; thin = 1;
total post-warmup draws = 1000
Multilevel Hyperparameters:
~animal (Number of levels: 1084)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sd(sex1) 1.32 0.24 0.81 1.75 1.02 40 124
sd(sex2) 0.91 0.38 0.12 1.53 1.07 22 70
~byear (Number of levels: 34)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sd(sex1) 0.91 0.17 0.62 1.26 1.00 632 604
sd(sex2) 1.08 0.20 0.76 1.50 1.00 429 614
~mother (Number of levels: 429)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sd(sex1) 0.91 0.24 0.33 1.33 1.03 93 93
sd(sex2) 1.39 0.16 1.09 1.69 1.02 210 262
Regression Coefficients:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept 6.28 0.23 5.85 6.74 1.00 635 490
sex2 2.05 0.34 1.39 2.67 1.00 636 698
Further Distributional Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 1.49 0.17 1.15 1.76 1.05 22 53
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
We can see the model estimate variance for both sexes. However, the residual level or sigma is not splitted by sexes. A futher and more complex code need to be performed, thus we can estimate the sex-specific heritability.
bf_m1.5<-bf(bwt~1+sex+(0+sex||gr(animal, cov =Amat))+(0+sex||mother)+(0+sex||byear),sigma~sex-1)brms_m1.5<-brm(bf_m1.5, data =gryphon, data2 =list(Amat =Amat), family =gaussian(), chains =1, cores =1, iter =1000)save(brms_m1.5, file ="r-obj/brms_m1_5.rda")
To save time, the results of the calculation is stored in the spare file brms_m1_4.rda".
Warning: Parts of the model have not converged (some Rhats are > 1.05). Be
careful when analysing the results! We recommend running more iterations and/or
setting stronger priors.
Family: gaussian
Links: mu = identity; sigma = log
Formula: bwt ~ 1 + sex + (0 + sex || gr(animal, cov = Amat)) + (0 + sex || mother) + (0 + sex || byear)
sigma ~ sex - 1
Data: gryphon (Number of observations: 854)
Draws: 1 chains, each with iter = 1000; warmup = 500; thin = 1;
total post-warmup draws = 500
Multilevel Hyperparameters:
~animal (Number of levels: 854)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sd(sex1) 1.56 0.29 1.02 2.09 1.17 4 30
sd(sex2) 1.61 0.41 0.52 2.08 1.36 2 21
~byear (Number of levels: 34)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sd(sex1) 0.91 0.18 0.59 1.36 1.01 153 229
sd(sex2) 1.06 0.20 0.75 1.49 1.00 170 143
~mother (Number of levels: 394)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sd(sex1) 0.88 0.21 0.41 1.25 1.01 73 134
sd(sex2) 1.27 0.18 0.88 1.59 1.01 31 64
Regression Coefficients:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept 6.29 0.23 5.88 6.75 1.00 209 313
sex2 2.02 0.31 1.49 2.66 1.00 127 296
sigma_sex1 0.22 0.21 -0.25 0.54 1.15 5 12
sigma_sex2 -0.20 0.40 -0.82 0.54 1.59 2 15
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
Iterations = 1:500
Thinning interval = 1
Number of chains = 1
Sample size per chain = 500
1. Empirical mean and standard deviation for each variable,
plus standard error of the mean:
Mean SD Naive SE Time-series SE
0.575443 0.126621 0.005663 0.031251
2. Quantiles for each variable:
2.5% 25% 50% 75% 97.5%
0.3075 0.4863 0.5811 0.6741 0.7800
Iterations = 1:500
Thinning interval = 1
Number of chains = 1
Sample size per chain = 500
1. Empirical mean and standard deviation for each variable,
plus standard error of the mean:
Mean SD Naive SE Time-series SE
0.463879 0.155395 0.006949 0.078323
2. Quantiles for each variable:
2.5% 25% 50% 75% 97.5%
0.06693 0.43668 0.50150 0.55729 0.66016
Unfortunately (to our knowledge), it is not possible to alter the variance matrices and refit them within the model.
4.0.7 Covariance between two random effects
Some research questions require to estimate the covariance between two random effects within a univariate model. Unfortunately (to our knowledge), it is not possible to create a covariance between distinct random effects (https://github.com/paul-buerkner/brms/issues/502). However,a multi-membership model can be fit using the linking.function mm, thus forcing the variance of two variables to be equal and the covariance to 1.
Wilson, A. J., D. Réale, M. N. Clements, M. M. Morrissey, E. Postma, C. A. Walling, L. E. B. Kruuk, et D. H. Nussey. 2010. An Ecologist’s Guide to the Animal Model. Journal of Animal Ecology 79:13‑26.